Chemical Education Journal (CEJ),
Vol. 12, No. 1 /Registration No. 12-2 /Received August 1, 2008.
URL = http://chem.sci.utsunomiya-u.ac.jp/cejrnlE.html
ABSTRACT
Both structure and reactivity are involved in the study of reaction
intermediates. The main types of intermediates of interest are
carbocations, carbanions, free radicals, and carbenes. Usually,
these intermediates are not isolable. Physical organic chemistry
is the study of the interrelationships between structure and reactivity
in organic molecules. Physical organic chemistry is the
study of organic chemistry using methods of physical chemistry
such as chemical equilibrium, chemical kinetics, thermochemistry,
and quantum chemistry. In the present article an attempt is made
to correlate the nature of heterolysis five different free radical
type nitroxyl adducts using Hammett's Liner Free Energy Relationships.
A linear correlation exists between the logarithm of a rate constant or equilibrium constant for one series of reactions and the logarithm of the rate constant or equilibrium constant for a related series of reactions respectively. Typical examples of such relations (also known as linear Gibbs energy relations) are the Brønsted relation and the Hammett equation. The name arises because the logarithm of an equilibrium constant (at constant temperature and pressure) is proportional to a standard free energy (Gibbs energy) change, and the logarithm of a rate constant is a linear function of the free energy (Gibbs energy) of activation. It has been suggested that this name should be replaced by linear Gibbs energy relation, but at present there is little sign of acceptance of this change. The area of Physical Organic Chemistry which deals with such relations is commonly referred to as 'Linear Free-Energy Relationships (LFER)'.
Gibbs energy of activation (standard free energy of activation)
is DGo![]() the standard Gibbs energy difference
between the transition state of a reaction (either an elementary
reaction or a stepwise reaction) and the ground state
of the reactants. It is calculated from
the experimental rate constant k via the conventional form
of the absolute rate equation: DG` = RT [ln (kB / h)
- ln (k /T)] where kB is the Boltzmann constant
and h the Planck constant (kB/h = 2.08358 ×
1010 K-1 s-1). The values of
the rate constants, and hence Gibbs energies of activation, depend
upon the choice of concentration units (or of the thermodynamic
standard state).
the standard Gibbs energy difference
between the transition state of a reaction (either an elementary
reaction or a stepwise reaction) and the ground state
of the reactants. It is calculated from
the experimental rate constant k via the conventional form
of the absolute rate equation: DG` = RT [ln (kB / h)
- ln (k /T)] where kB is the Boltzmann constant
and h the Planck constant (kB/h = 2.08358 ×
1010 K-1 s-1). The values of
the rate constants, and hence Gibbs energies of activation, depend
upon the choice of concentration units (or of the thermodynamic
standard state).
Although spontaneous transformations
all have negative DGºo
s, not all exergonic processes are spontaneous, due to activation
energy barriers to reaction. Treatment of activation energy was
framed in terms of enthalpy or potential energy. It should now
be clear that, if entropy factors are to be incorporated in the
activation barrier, we should be thinking about Free Energy
of Activation, DG![]() . The defining equation then becomes:
. The defining equation then becomes:
DG![]() = DH
= DH ![]() -
T DS
-
T DS![]()
Transition state theory proposes an equilibrium between reactants
and the transition state, so each of the functions in this equation
may be viewed as a [FTransition
State - FReactants]
difference, where F represents H, S or G.
The equations demonstrate the similar exponential relationship
of DGºº to Keq and DG![]() to k.
to k.
| and | 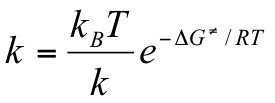 |
Since the rate constant equation incorporates the temperature
variable twice and DG![]() also changes with temperature, observed
reaction rates are clearly temperature dependent. Physical Organic
Chemists make general use of this relationship in two ways. First,
it is a rule-of-thumb that a 10º C increase in reaction temperature
will roughly double the rate of that reaction. Second, since this
rule applies as well to accompanying reactions, the rates of such
side reactions also increase with temperature, sometimes more
than the desired reaction. Consequently, the practical yield of
the desired product may actually decrease at higher temperatures.
Thus, a cleaner (less contaminated) product is often obtained
by running a reaction at the lowest effective temperature that
gives the desired product. Because DG
also changes with temperature, observed
reaction rates are clearly temperature dependent. Physical Organic
Chemists make general use of this relationship in two ways. First,
it is a rule-of-thumb that a 10º C increase in reaction temperature
will roughly double the rate of that reaction. Second, since this
rule applies as well to accompanying reactions, the rates of such
side reactions also increase with temperature, sometimes more
than the desired reaction. Consequently, the practical yield of
the desired product may actually decrease at higher temperatures.
Thus, a cleaner (less contaminated) product is often obtained
by running a reaction at the lowest effective temperature that
gives the desired product. Because DG ![]() incorporates
a temperature dependent entropy factor and is related exponentially
to the rate constant, k, reaction rate studies at different
temperatures may be interpreted to provide the activation parameters.
incorporates
a temperature dependent entropy factor and is related exponentially
to the rate constant, k, reaction rate studies at different
temperatures may be interpreted to provide the activation parameters.
Both the Arrhenius and the Eyring equation describe the temperature dependence of reaction rate. Strictly speaking, the Arrhenius equation can be applied only to gas reactions. The Eyring equation is used in the study of gas, condensed and mixed phase reactions - all places where the simple collision model is not very helpful. The Arrhenius equation is founded on the empirical observation that conducting a reaction at a higher temperature increases the reaction rate. The Eyring equation is a theoretical construct, based on transition state model.
Now let us derive a LFER on the basis of Eyring and Hammett
equations: Taking Eyring equation
.jpg) .........(1)
.........(1)
Taking logarithm
.jpg) ......... (2)
......... (2)
and from Hammett equation
log k = log ko + rs .........(3)
from eqn. 2 and 3
![]() ......... (4)
......... (4)
Simplifying and rearranging eqn. 4 one will get
DG ![]() =
= ![]() - 2.303RTrs ... (5)
- 2.303RTrs ... (5)
This equation with a particular value of r, applies to any
reaction involving a reactant having a series of substituents.
For a second series of homologous reactions, with reaction constant
r',
one can show that
DG ![]() '=
'= ![]() - 2.303 RT r's
.........(6)
- 2.303 RT r's
.........(6)
Dividing the equation 5 and 6 by r and r'
respectively and subtracting one from the other and on simplification,
we get
DG![]() = (r/r') DG
= (r/r') DG ![]() '+ constant.... (7)
'+ constant.... (7)
Thus there is a linear relationship between DG ![]() of one series
and to DG
of one series
and to DG![]() ' of another series. Now let us see the application
of this relationship to the following putative examples.
' of another series. Now let us see the application
of this relationship to the following putative examples.
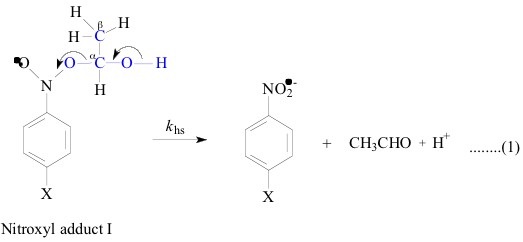
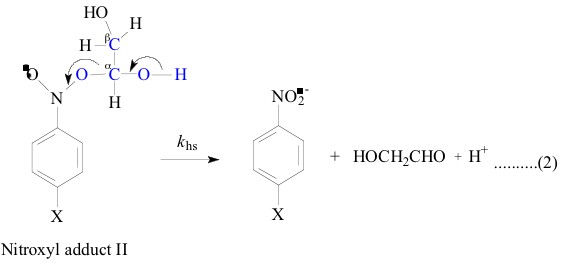
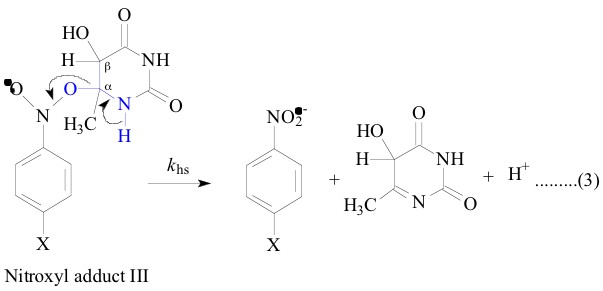
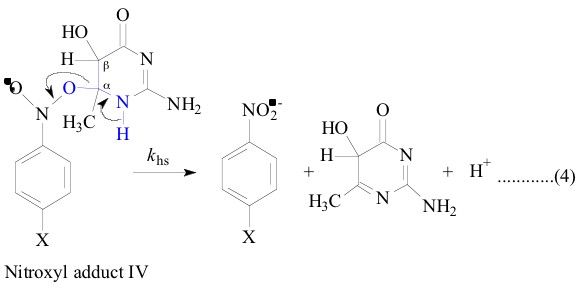
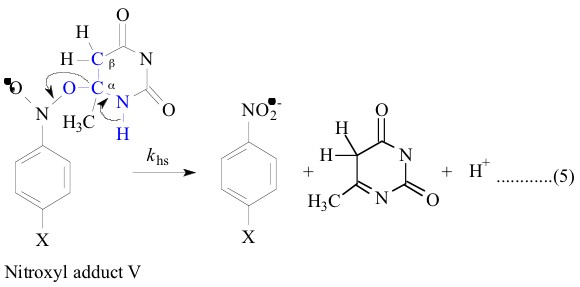
All the nitroxyl adducts I-V were generated [1-4]
pulseradiolytically in aqueous solution from nitrobenzenes and
the corresponding a-hydroxyethyl, a,b- dihydroxyethyl,
6-methyluracil-6-yl, 6-methyl-i-cytosine-6-yl and 6-methyldihydrouracil-6-yl
radicals. Production of a-hydroxyethyl, a,b-dihydroxyethyl,
6-methyluracil-6-yl, 6-methyl-i-cytosine-6-yl and 6-methyldihydrouracil-6-yl
radicals were described else where [1-4]. The heterolysis rate constants, khs, and
all activation free energy changes were taken from those references.
The LFER plot for the heterolysis reaction of the nitroxyl adducts
I and V is linear (Figure 1; r =
0.99). The slope of this plot, which should be equal to the ratio
of the Hammett ¡ values for the heterolysis of the two,
adducts I and V, results as 1.0, in agreement with the individually
determined ¡ values (both 1.5). Also the solvent isotope
effect (SIE) on khs of adduct V, that is khs(H2O)/ khs(D2O) was
found to be 1.7 - 2.3 is similar to that of 2.2 of the adduct
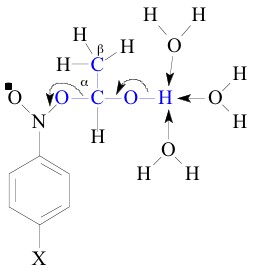
I. All these observations led to conclude that the two transition
states of the heterolysis of adduct I and adduct V were essentially
similar in terms of synchronous O-H and C-O bond breaking assisted
by solvent water as shown below. This leads to immobilization
of solvent water which was reflected in high negative DS`![]() [1, 4].
[1, 4].
 |
and | 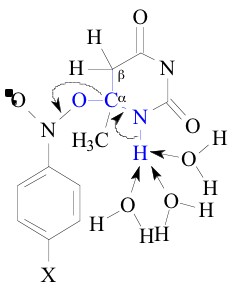 |
Hence these are all plenty of evidences for the close resemblance of the ±-hydroxyethyl (CH3C.HOH) and 6-methyldihydrouracil-6-yl radicals and which lead to the similar transition states of the adducts on their way to products.
In contrast, the LFER plot for the heterolysis
reaction of the nitroxyl adducts III and IV were not perfectly
linear when plotted against the nitroxyl adduct I and adduct V
(Figure
2, Figure 3, Figure
5 and Figure 6; r =
only 0.93, 0.92, 0.95 and 0.91 respectively) which indicates that
the ±-hydroxyethyl (CH3C.HOH) is not a good model for the 6-methyluracil-6-yl
and 6-methyl-i-cytosine-6-yl radicals. That these belong
to a class of their own is shown not only by similar solvent KIE
(1.2 - 1.4) which is similar to that of (1.5) observed for the
heterolysis of the adduct II but also by the good correlation
(Figure
4; r = 0.98) between their own (for
the heterolysis of nitroxyl adducts III and IV) corresponding
DG`![]() values.
To make an attempt to correlate the DG
values.
To make an attempt to correlate the DG![]() ` values of adduct III and IV with that of adduct
II, sufficient data for adduct II is not available. Yet the similar
solvent KIE is a strong evidence to say that the a,b-dihydroxyethyl
radical (HO.CHCH2OH) is a good model for the 6-methyluracil-6-yl
and 6-methyl-i-cytosine-6-yl radicals when their adducts,
III and IV lead to similar transition states on their way to the
products. And adducts II, III and IV differ with adducts I and
V where former set have an OH group in b position as shown
in their structures above. The strong difference in KIE between
the two sets of adducts I, V (1.7 - 2.3) and II, III, IV (1.2
- 1.5) could be understood in terms of the differences in stabilities
between the transition states of the two different sets of adducts.
The two sets of transition states differ by an OH group in b
position to the deprotonatable OH/NH group. Due to the -I effect
of OH group in the b position, the adducts II, III and IV are expected
to be less stable than the adducts I and V. Besides in the reduction
of khs (of C-O bond heterolysis, which is measured
in terms of the build up kinetics of the nitro benzene radical
anion), this leads to an increased tendency to deprotonate at
an early stage of the transition state, i.e. deprotonation tends
to precede the heterolysis of C-O bond. This is symbolized as
shown below:
` values of adduct III and IV with that of adduct
II, sufficient data for adduct II is not available. Yet the similar
solvent KIE is a strong evidence to say that the a,b-dihydroxyethyl
radical (HO.CHCH2OH) is a good model for the 6-methyluracil-6-yl
and 6-methyl-i-cytosine-6-yl radicals when their adducts,
III and IV lead to similar transition states on their way to the
products. And adducts II, III and IV differ with adducts I and
V where former set have an OH group in b position as shown
in their structures above. The strong difference in KIE between
the two sets of adducts I, V (1.7 - 2.3) and II, III, IV (1.2
- 1.5) could be understood in terms of the differences in stabilities
between the transition states of the two different sets of adducts.
The two sets of transition states differ by an OH group in b
position to the deprotonatable OH/NH group. Due to the -I effect
of OH group in the b position, the adducts II, III and IV are expected
to be less stable than the adducts I and V. Besides in the reduction
of khs (of C-O bond heterolysis, which is measured
in terms of the build up kinetics of the nitro benzene radical
anion), this leads to an increased tendency to deprotonate at
an early stage of the transition state, i.e. deprotonation tends
to precede the heterolysis of C-O bond. This is symbolized as
shown below:
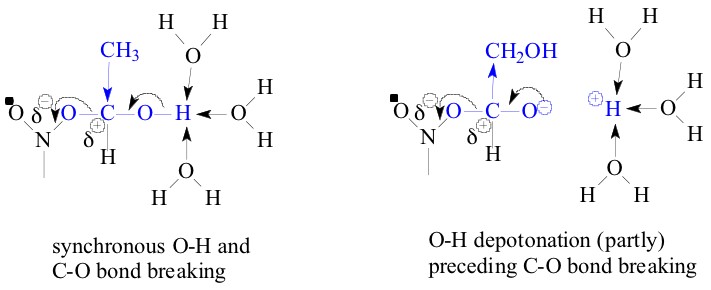
So in conclusion the two sets of adducts (I and V), and (II, III and IV) behave differently and one can conclude that the a-hydroxyethyl radical is a good model for 6-methyldihydrouracil-6-yl radical and a,b-dihydroxyethyl radical is a good model for 6-methyluracil-6-yl and 6-methyl-i-cytosine-6-yl radicals. The structures of the radical anions in the steps 1 - 6 could be read as the following:
Dedication: This article is dedicated to my mentor Prof. Dr. Steen Steenken (Retd), Max-Planck-Institute for Radiation Chemistry, Muelheim a.d. Ruhr, Germany.
References:
[1]
Jagannadham, V. and Steenken, S. (1984), J. Am. Chem. Soc.
106: 6542-6551.
[2] Steenken,
S. and Jagannadham, V. (1985), J. Am. Chem. Soc. 107:
6818-6826.
[3] Jagannadham,
V. and Steenken, S. (1988), J. Phys. Chem. 92: 111-118.
[4] Jagannadham,
V. and Steenken, S. (1988), J. Am. Chem. Soc. 110:
2188-2192.