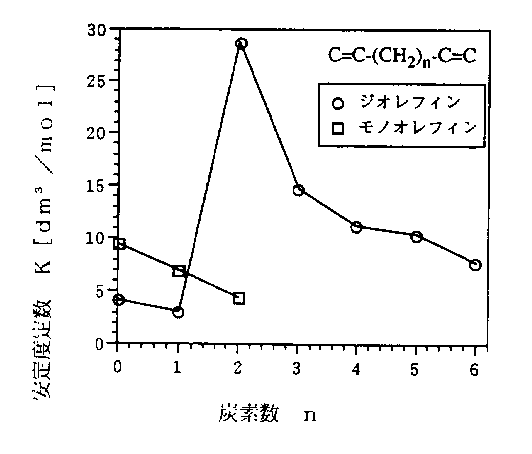
丂丂丂昞俀 僆儗僼傿儞暘棧朄[2] ----------------------------------------------- 婛懚暘棧朄丗掅壏忲棷朄丄拪弌忲棷朄 暔棟媧拝朄 怴婯暘棧朄丗壔妛媧拝朄(Cu(I)扴帩庽帀偺棙梡乯 壔妛媧廂朄(Ag(I),Cu(I)悈梟塼乯 Ag(I) 嶖懱宆懀恑奼嶶棙梡枌暘棧朄 -----------------------------------------------
戞堦摵傪僆儗僼傿儞暘棧偵棙梡偡傞婛墲偺帋傒傪昞俁[2]偵傑偲傔偨丅戞堦摵墫偺悈梟塼傪梡偄偰僈僗忬僆儗僼傿儞傪媧廂暘棧偡傞曽朄[4]偱偼戞堦摵墫偑悈偵梟偗偢傜偄寚揰偑偁傞丅傾儞儌僯傾傗墫慺僀僆儞傪揧壛偟偰戞堦摵墫偺梟夝惈傪崅傔傞偙偲傕偱偒傞偑丄偙傟傜偑摵偵桪愭揑偵攝埵偡傞偨傔偵僆儗僼傿儞夞廂梕検偺掅壓傪傕偨傜偡丅僼儖僆儘壔崌暔傗擇嬥懏墫偵傛偭偰梟夝惈傪崅傔傞偙偲傕壜擻偱偁傞偑丄偙傟傜帺恎偵暘夝惈偑偁偭偰埨掕偱偼側偄[5,6]丅偦偙偱丄巁壔丒娨尦斀墳偵傛偭偰戞堦摵傪惗惉偝偣傞偲摨帪偵僆儗僼傿儞偲兾嶖懱傪宍惉偝偣傞曽朄偑採埬偝傟偨丅偟偐偟丄嬥懏摵偺巁壔朄偼夁墫慺巁偺棙梡偵傛偭偰傾僙僠儗儞側偳偺敋敪惈壔崌暔偺惗惉傪堷偒婲偙偡偺偱岲傑偟偔側偄丅
昞俁 Cu(I) 偺僆儗僼傿儞暘棧傊偺棙梡[2]
-----------------------------------------------
曽朄 丂丂 寚揰
-----------------------------------------------
侾丏戞堦摵墫悈梟塼
(1)CuCl 丂丂丂丂丂丂丂丂擄梟惈丄晄嬒堦宯
(2)NH3,Cl-揧壛丂丂丂丂丂桪愭攝埵偵傛傞梕検掅壓
(3)(CF3SO4)CuB,丂丂丂丂 暘夝惈偑偁傝晄埨掕
CuAlX4
俀丏嬥懏摵偺巁壔
(1) 夁墫慺巁悈梟塼丂丂丂敋敪惈丄晄嬒堦宯
俁丏Cu(0)/Cu(I) 愙怗宯丂丂晄嬒堦宯
(1) 墫巁巁惈壓偱Cu(0)/Cu(I) 傪愙怗
(2) 僆儗僼傿儞懚嵼壓偱Cu(0)/Cu(II)傪愙怗
------------------------------------------------
墫巁懚嵼壓偱戞擇摵偲嬥懏摵傪愙怗偝偣傞曽朄偼墫壔戞堦摵偺惗惉朄偲偟偰抦傜傟偰偄傞丅傑偨丄僆儗僼傿儞懚嵼壓偱嬥懏摵偲戞擇摵傪愙怗偝偣偰屳偄偵巁壔丒娨尦偟偰兾嶖懱傪宍惉偡傞嫽枴偁傞曽朄偼岞奐摿嫋[7]偵偍偄偰採埬偝傟偰偄傞丅偙偺曽朄偼C4棷暘偺媧廂暘棧偵懳偟偰偦偺桳岠惈偑妋擣偝傟偰偄傞丅塼忬僆儗僼傿儞偺暘棧偱偼儊僞僲乕儖傪梟攠偲偡傞僗僠儗儞乛僄僠儖儀儞僛儞偺拪弌暘棧偵梡偄偨堦椺偺傒偑柧傜偐偵偝傟偰偄傞[8]丅
係丏俠倳乮俬乯偵傛傞侾丄俆僿僉僒僕僄儞偺拪弌暘棧[2]
丂俙倗乮俬乯偵懳偟偰崅偄嶖宍惉擻傪帵偡侾丆俆亅僿僉僒僕僄儞傪儌僨儖晄朞榓壔崌暔偲偟偰嬥懏摵乛戞擇摵悈梟塼宯傪拪弌暘棧嵻偵梡偄傞壜擻惈傪専摙偟偨丅
撪宎70mm偺奾漚憛偵1M-Cu(NO3)2傪100 dm3偲丄塼忬扽壔悈慺崿崌暔[50dm3,1,5-僿僉僒僕僄儞(10wt%)亄1-僿僉僙儞(15wt%)亄僔僋儘僿僉僙儞(20wt%)亄僩儖僄儞(15wt%)+n- 僿僾僞儞(40wt%)]偍傛傃嬥懏摵暡枛(悈梟塼拞偱1mol/dm3)傪壛偊偰丄俇枃暯塇崻僞乕價儞宆梼偱奾漚偟偨丅扽壔悈慺憡傪僈僗僋儘暘愅偟偰晄朞榓壔崌暔偺尭彮検傪媮傔偰拪弌検傪寛掕偟偨丅
係丏侾 娤嶡寢壥偲拪弌婡峔[2]
Cu(NO3)2偼悈偵梟偗偰Cu(II)僀僆儞偺惵怓傪掓偡傞偑丄僆儗僼傿儞偲斀墳偟偰Cu(I)- 僆儗僼傿儞兾嶖懱傪宍惉偡傞偲悈憡偼Cu(I) 僀僆儞偺椢怓傪掓偡傞丅摿偵丄奾漚梼愭抂嬤朤偱偼嫮偄椢怓偺棳懱夠偑尒傜傟偨丅
丂奾漚慜偺摵暡枛乮暯嬒宎偼栺0.2mm)偼桘悈奅柺偵懡偔偑懲愊偟偰偄偨丅奾漚掆巭屻偺崌堦奅柺偺堏摦曽岦偑壓岦偒偱偁傞偙偲偐傜奾漚拞偺暘嶶偺宆偼俷乛倂宆偱偁傞偙偲偑傢偐傞丅
丂娤嶡寢壥偐傜恾俀偺傛偆側拪弌婡峔偑峫偊傜傟傞丅僆儗僼傿儞偼桘憡偐傜悈憡偵暘攝偡傞丅悈憡偺奅柺嬤朤偱嬥懏摵偲Cu(II)偑愙怗偟偰晄埨掕側Cu(I) 偑惗惉偡傞丅潣漚梼偺愭抂椞堟偱偼棳懱偺棎傟偑嫮偄偺偱Cu(I) 偺惗惉懍搙偑崅偔丄椢怓傪掓偡傞丅偙偺Cu(I) 偺堦晹偵僆儗僼傿儞偑攝埵偟偰埨掕側嶖懱偑宍惉偝傟傞丅嶖懱偼悈梟塼傪椢怓偵曄偊傞丅
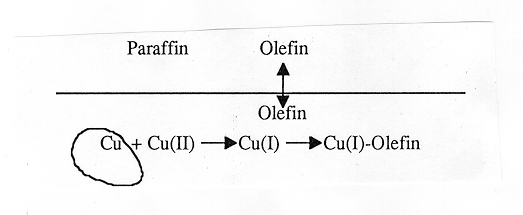
恾俀丂俠倳乮侽乯乛俠倳乮俬俬乯愙怗宯偵偍偗傞僆儗僼傿儞偺拪弌婡峔
係丏俀丂拪弌懍搙[2]
恾俁偵丄侾丆俆僿僉僒僕僄儞偺拪弌棪偺宱帪曄壔偵媦傏偡奾漚懍搙偺塭嬁傪帵偡丅偙偺拪弌宯偼摵暡枛傪娷傓晄嬒堦斀墳宯偱偁傞偺偱斀墳惉暘傗嶖懱偺奼嶶偑拪弌懍搙偵嫮偔塭嬁偡傞偙偲傪帵偟偰偄傞丅偙偺揰偼Cu(0)/Cu(II)愙怗拪弌朄偺寚揰偲偟偰嫇偘傜傟傞丅
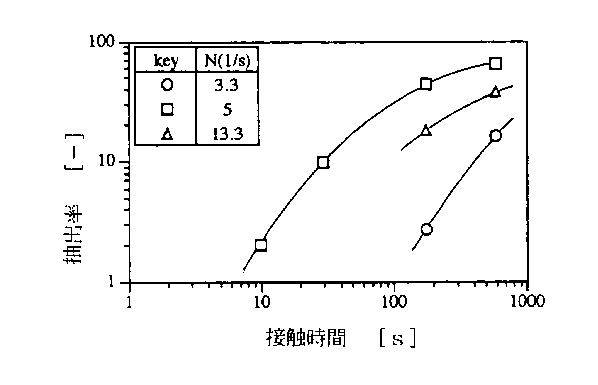
恾俁丂侾丄俆僿僉僒僕僄儞偺拪弌棪偺宱帪曄壔偵媦傏偡奾漚懍搙偺塭嬁[2]
丂恾係偵侾丄俆僿僉僒僕僄儞偲侾亅僿僉僙儞偺拪弌棪偺宱帪曄壔傪斾妑偟偨丅恾係偵偼AgNO3 悈梟塼傪梡偄偨応崌偺拪弌嬋慄傕帵偟偰偁傞偑丄偙偺応崌偵偼嬒堦宯偱偁傞偐傜奼嶶偺掞峈偑彫偝偄偺偱拪弌懍搙偼嬌傔偰懍偄丅
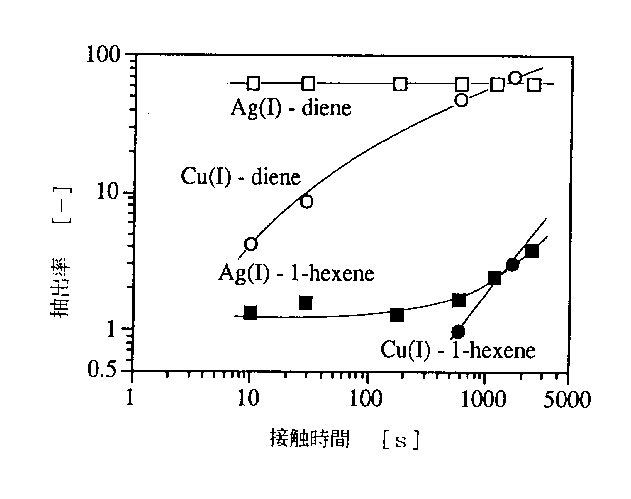
丂恾係丂俠倳乮侽乯乛俠倳乮俬俬乯愙怗拪弌宯偲徤巁嬧悈梟塼偵傛傞僆儗僼傿儞偺拪弌棪偺宱帪曄壔[2]
係丏俁 暯峵拪弌検[2]
恾係偺Cu(0)/Cu(II)拪弌宯偺寢壥偱偼拪弌暯峵偵偼摓払偟偰偄側偄偑丄廫暘偵愙怗偣傞偲俙倗乮俬乯嶖懱偵斾傋偰暯峵拪弌検偑崅偔側傞孹岦傪帵偟偰偄傞丅偙偺寢壥偼俙倗乮俬乯偵斾傋偰俠倳乮俬乯偺曽偑嶖宍惉掕悢偼戝偒偄偲偄偆堦斒揑孹岦[1]偵崌抳偟偰偄傞丅
丂恾係傪傒傞偲丄俠倳乮俬乯偵偍偄偰傕俙倗乮俬乯偲摨條偵丄侾丄俆僿僉僒僕僄儞偺曽偑侾亅僿僉僙儞傛傝暯峵拪弌検偼挊偟偔戝偒偄偺偱丄俠倳乮俬乯亅侾丄俆僿僉僒僕僄儞偺僉儗乕僩傪宍惉偟偰偄傞壜擻惈偑崅偄丅幚嵺偵丄俠倳乮俬乯偲侾丆俆僿僉僒僕僄儞偺寢崌斾棪偼侾丗侾偱偁傞偙偲偑柧傜偐偵偝傟偰偄傞[9]丅僩儖僄儞側偳懠偺惉暘偵偮偄偰偼暘愅惛搙撪偱拪弌偝傟偨挍岓偼尒傜傟側偐偭偨丅
俆丏擕壔塼枌暘棧朄傊偺墳梡
丂俠倳乮俬乯僀僆儞傪娷傓倧乛倵乛倧擕壔塼枌朄偵傛偭偰侾丆俆亅僿僉僒僕僄儞傪暘棧偡傞偲憐掕偟偰丄婛懚偺擕壔塼枌摟夁僨乕僞偵擣傔傜傟傞娭學偐傜拪弌懍搙傪悇掕偟偰傒傞丅
丂Kato傜[10]偼丄婛墲偺曬崘偵偁傞擕壔塼枌摟夁僨乕僞傪梡偄偰憤妵摟夁梕検學悢俲i倎傪媮傔丄奼嶶棩懍偺忬懺偱偁傟偽俲i倎偲暘攝斾俢i偺娫偵師幃偺憡娭娭學偑惉傝棫偮偙偲傪尒偄偩偟偰偄傞丅
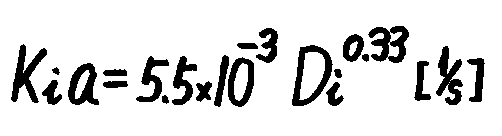
偙偺娭學傪恾俆偵帵偡丅堦曽丄Cu(0)/1M-Cu(NO3)2宯偵傛偭偰侾丄俆僿僉僒僕僄儞傪拪弌偡傞嵺偺暘攝斾偼侽丏俁 掱搙偱偁傞偐傜[9]丄恾俆忋偱乮侾乯幃傪枮偨偡擕壔塼枌摟夁僨乕僞偲斾傋傞偲丄俢俀俤俫俹俙側偳偵傛傞媯傒忋偘岠壥傪棙梡偡傞傾儈僲巁偲摨偠掱搙偺摟夁懍搙傪帵偡偲梊憐偝傟傞丅偨偩偟丄廫暘偵斀墳懍搙傪崅傔偰嬒堦斀墳宯偵偍偗傞奼嶶棩懍忬懺偑幚尰偝傟傞偲壖掕偟偰偄傞丅側偍丄乮侾乯幃偺娭學偼暘嶶偺宆(w/o/w,o/w/o)偵埶懚偣偢丄崅偄摟夁懍搙傪幚尰偡傞憖嶌忦審偱偁偭偰奼嶶棩懍忬懺偵偁傟偽亇俆侽亾偺惛搙撪偱擣傔傜傟傞.傑偨丄拞嬻巺儌僕儏乕儖[11]偵斾傋偰傕擕壔塼枌偺梕検學悢偼崅偔丄僄儅儖僔儑儞揌宎乛撪晹塼揌宎偺斾傪侾偵嬤偯偗傞偙偲偵傛偭偰暘棧學悢傪偨偐傔傜傟傞[12,13]偙偲偑帵偝傟偰偄傞丅
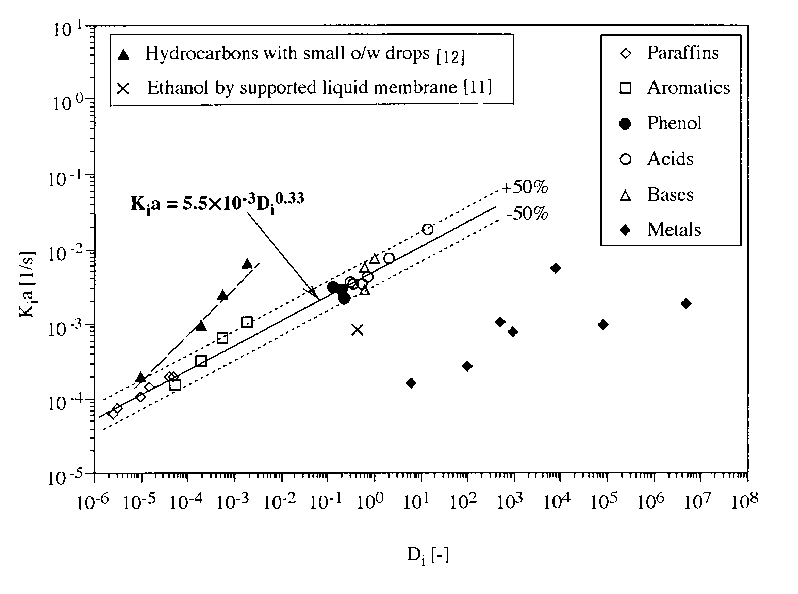
丂丂丂恾俆丂擕壔塼枌朄偺憤妵摟夁梕検學悢偲暘攝斾偺娭學[10]
俇丏偍傢傝偵
C4僆儗僼傿儞偺媧廂暘棧偵偍偄偰桳岠惈偑妋擣偝傟偰偄傞嬥懏摵乛戞擇摵梟塼愙怗暘棧宯傪塼忬僆儗僼傿儞偺拪弌暘棧偵棙梡偡傞偨傔偵丄堦憌専摙傪恑傔傞昁梫偑偁傞傕偺偲峫偊傜傟傞丅偙偺曽朄偼嬥懏摵傪娷桳偡傞晄嬒堦斀墳宯偲偄偆寚揰偼偁傞偑丄楑壙偱兾嶖懱宍惉擻偑崅偄偲偄偆摵偺摿挿傪妶梡偡傞偱偒傞傕偺偲婜懸偝傟傞丅 堦曽丄斀墳惈偺崅偄摵傪梡偄傞偙偲偵傛傞晄棙塿偵偮偄偰傕専摙傪恑傔側偗傟偽側傜側偄丅偡側傢偪丄拪弌憖嶌偵傛傞僆儗僼傿儞偺曄幙偵偮偄偰挷傋丄幚嵺偵媡拪弌偑壜擻偱偁傞偙偲傕妋擣偟側偗傟偽側傜側偄丅
堷梡暥專
1) Hartley,F.H.: Chem.Reviews, 73,163(1973)
2) 壛摗丂妎,働儈僇儖僄儞僕僯傾儕儞僌,40,27(1995).
3) Eldridge,H.B., IEC.Res.,32,2208(1993)
4) Gilliland et al., J.A.C.S.,63,2088(1941)
5) 摿奐丄徍51-26806
6) 摿岞丄徍48-35041
7) 摿奐丄徍50-18425
8) 摿奐丄徍57-58633
9) Kato,S.,K.Nakano,H.Noritomi,K.Nagahama:Solv.Ext.Res.Dev.,Japan,3,117(1996).
10)Kato,S.,K.Nakano,H.Noritomi,K.Nagahama:Solv.Ext.Res.Dev.,Japan, in press.
11)Vatai,G.,M.N.Tekic:Sep.Sci.Tech.,26,1005(1991).
12)Kato,S.,J.Kawasaki,J.Chem.Eng.Japan,20,140(1987).
13)Egashira,R.H.Tanno,S.Kato,J.Kawasaki,J.Chem.Eng.Japan,28,38(1995).
昞俁 Cu(I) 偺僆儗僼傿儞暘棧傊偺棙梡[2] ----------------------------------------------- 曽朄 丂丂 寚揰 ----------------------------------------------- 侾丏戞堦摵墫悈梟塼 (1)CuCl 丂丂丂丂丂丂丂丂擄梟惈丄晄嬒堦宯 (2)NH3,Cl-揧壛丂丂丂丂丂桪愭攝埵偵傛傞梕検掅壓 (3)(CF3SO4)CuB,丂丂丂丂 暘夝惈偑偁傝晄埨掕 CuAlX4 俀丏嬥懏摵偺巁壔 (1) 夁墫慺巁悈梟塼丂丂丂敋敪惈丄晄嬒堦宯 俁丏Cu(0)/Cu(I) 愙怗宯丂丂晄嬒堦宯 (1) 墫巁巁惈壓偱Cu(0)/Cu(I) 傪愙怗 (2) 僆儗僼傿儞懚嵼壓偱Cu(0)/Cu(II)傪愙怗 ------------------------------------------------
係丏俠倳乮俬乯偵傛傞侾丄俆僿僉僒僕僄儞偺拪弌暘棧[2]
丂俙倗乮俬乯偵懳偟偰崅偄嶖宍惉擻傪帵偡侾丆俆亅僿僉僒僕僄儞傪儌僨儖晄朞榓壔崌暔偲偟偰嬥懏摵乛戞擇摵悈梟塼宯傪拪弌暘棧嵻偵梡偄傞壜擻惈傪専摙偟偨丅
撪宎70mm偺奾漚憛偵1M-Cu(NO3)2傪100 dm3偲丄塼忬扽壔悈慺崿崌暔[50dm3,1,5-僿僉僒僕僄儞(10wt%)亄1-僿僉僙儞(15wt%)亄僔僋儘僿僉僙儞(20wt%)亄僩儖僄儞(15wt%)+n- 僿僾僞儞(40wt%)]偍傛傃嬥懏摵暡枛(悈梟塼拞偱1mol/dm3)傪壛偊偰丄俇枃暯塇崻僞乕價儞宆梼偱奾漚偟偨丅扽壔悈慺憡傪僈僗僋儘暘愅偟偰晄朞榓壔崌暔偺尭彮検傪媮傔偰拪弌検傪寛掕偟偨丅
係丏侾 娤嶡寢壥偲拪弌婡峔[2]
Cu(NO3)2偼悈偵梟偗偰Cu(II)僀僆儞偺惵怓傪掓偡傞偑丄僆儗僼傿儞偲斀墳偟偰Cu(I)- 僆儗僼傿儞兾嶖懱傪宍惉偡傞偲悈憡偼Cu(I) 僀僆儞偺椢怓傪掓偡傞丅摿偵丄奾漚梼愭抂嬤朤偱偼嫮偄椢怓偺棳懱夠偑尒傜傟偨丅
丂奾漚慜偺摵暡枛乮暯嬒宎偼栺0.2mm)偼桘悈奅柺偵懡偔偑懲愊偟偰偄偨丅奾漚掆巭屻偺崌堦奅柺偺堏摦曽岦偑壓岦偒偱偁傞偙偲偐傜奾漚拞偺暘嶶偺宆偼俷乛倂宆偱偁傞偙偲偑傢偐傞丅
丂娤嶡寢壥偐傜恾俀偺傛偆側拪弌婡峔偑峫偊傜傟傞丅僆儗僼傿儞偼桘憡偐傜悈憡偵暘攝偡傞丅悈憡偺奅柺嬤朤偱嬥懏摵偲Cu(II)偑愙怗偟偰晄埨掕側Cu(I) 偑惗惉偡傞丅潣漚梼偺愭抂椞堟偱偼棳懱偺棎傟偑嫮偄偺偱Cu(I) 偺惗惉懍搙偑崅偔丄椢怓傪掓偡傞丅偙偺Cu(I) 偺堦晹偵僆儗僼傿儞偑攝埵偟偰埨掕側嶖懱偑宍惉偝傟傞丅嶖懱偼悈梟塼傪椢怓偵曄偊傞丅
恾俀丂俠倳乮侽乯乛俠倳乮俬俬乯愙怗宯偵偍偗傞僆儗僼傿儞偺拪弌婡峔
係丏俀丂拪弌懍搙[2]
恾俁偵丄侾丆俆僿僉僒僕僄儞偺拪弌棪偺宱帪曄壔偵媦傏偡奾漚懍搙偺塭嬁傪帵偡丅偙偺拪弌宯偼摵暡枛傪娷傓晄嬒堦斀墳宯偱偁傞偺偱斀墳惉暘傗嶖懱偺奼嶶偑拪弌懍搙偵嫮偔塭嬁偡傞偙偲傪帵偟偰偄傞丅偙偺揰偼Cu(0)/Cu(II)愙怗拪弌朄偺寚揰偲偟偰嫇偘傜傟傞丅
恾俁丂侾丄俆僿僉僒僕僄儞偺拪弌棪偺宱帪曄壔偵媦傏偡奾漚懍搙偺塭嬁[2]
丂恾係偵侾丄俆僿僉僒僕僄儞偲侾亅僿僉僙儞偺拪弌棪偺宱帪曄壔傪斾妑偟偨丅恾係偵偼AgNO3 悈梟塼傪梡偄偨応崌偺拪弌嬋慄傕帵偟偰偁傞偑丄偙偺応崌偵偼嬒堦宯偱偁傞偐傜奼嶶偺掞峈偑彫偝偄偺偱拪弌懍搙偼嬌傔偰懍偄丅
丂恾係丂俠倳乮侽乯乛俠倳乮俬俬乯愙怗拪弌宯偲徤巁嬧悈梟塼偵傛傞僆儗僼傿儞偺拪弌棪偺宱帪曄壔[2]
係丏俁 暯峵拪弌検[2]
恾係偺Cu(0)/Cu(II)拪弌宯偺寢壥偱偼拪弌暯峵偵偼摓払偟偰偄側偄偑丄廫暘偵愙怗偣傞偲俙倗乮俬乯嶖懱偵斾傋偰暯峵拪弌検偑崅偔側傞孹岦傪帵偟偰偄傞丅偙偺寢壥偼俙倗乮俬乯偵斾傋偰俠倳乮俬乯偺曽偑嶖宍惉掕悢偼戝偒偄偲偄偆堦斒揑孹岦[1]偵崌抳偟偰偄傞丅
丂恾係傪傒傞偲丄俠倳乮俬乯偵偍偄偰傕俙倗乮俬乯偲摨條偵丄侾丄俆僿僉僒僕僄儞偺曽偑侾亅僿僉僙儞傛傝暯峵拪弌検偼挊偟偔戝偒偄偺偱丄俠倳乮俬乯亅侾丄俆僿僉僒僕僄儞偺僉儗乕僩傪宍惉偟偰偄傞壜擻惈偑崅偄丅幚嵺偵丄俠倳乮俬乯偲侾丆俆僿僉僒僕僄儞偺寢崌斾棪偼侾丗侾偱偁傞偙偲偑柧傜偐偵偝傟偰偄傞[9]丅僩儖僄儞側偳懠偺惉暘偵偮偄偰偼暘愅惛搙撪偱拪弌偝傟偨挍岓偼尒傜傟側偐偭偨丅
俆丏擕壔塼枌暘棧朄傊偺墳梡
丂俠倳乮俬乯僀僆儞傪娷傓倧乛倵乛倧擕壔塼枌朄偵傛偭偰侾丆俆亅僿僉僒僕僄儞傪暘棧偡傞偲憐掕偟偰丄婛懚偺擕壔塼枌摟夁僨乕僞偵擣傔傜傟傞娭學偐傜拪弌懍搙傪悇掕偟偰傒傞丅
丂Kato傜[10]偼丄婛墲偺曬崘偵偁傞擕壔塼枌摟夁僨乕僞傪梡偄偰憤妵摟夁梕検學悢俲i倎傪媮傔丄奼嶶棩懍偺忬懺偱偁傟偽俲i倎偲暘攝斾俢i偺娫偵師幃偺憡娭娭學偑惉傝棫偮偙偲傪尒偄偩偟偰偄傞丅
偙偺娭學傪恾俆偵帵偡丅堦曽丄Cu(0)/1M-Cu(NO3)2宯偵傛偭偰侾丄俆僿僉僒僕僄儞傪拪弌偡傞嵺偺暘攝斾偼侽丏俁 掱搙偱偁傞偐傜[9]丄恾俆忋偱乮侾乯幃傪枮偨偡擕壔塼枌摟夁僨乕僞偲斾傋傞偲丄俢俀俤俫俹俙側偳偵傛傞媯傒忋偘岠壥傪棙梡偡傞傾儈僲巁偲摨偠掱搙偺摟夁懍搙傪帵偡偲梊憐偝傟傞丅偨偩偟丄廫暘偵斀墳懍搙傪崅傔偰嬒堦斀墳宯偵偍偗傞奼嶶棩懍忬懺偑幚尰偝傟傞偲壖掕偟偰偄傞丅側偍丄乮侾乯幃偺娭學偼暘嶶偺宆(w/o/w,o/w/o)偵埶懚偣偢丄崅偄摟夁懍搙傪幚尰偡傞憖嶌忦審偱偁偭偰奼嶶棩懍忬懺偵偁傟偽亇俆侽亾偺惛搙撪偱擣傔傜傟傞.傑偨丄拞嬻巺儌僕儏乕儖[11]偵斾傋偰傕擕壔塼枌偺梕検學悢偼崅偔丄僄儅儖僔儑儞揌宎乛撪晹塼揌宎偺斾傪侾偵嬤偯偗傞偙偲偵傛偭偰暘棧學悢傪偨偐傔傜傟傞[12,13]偙偲偑帵偝傟偰偄傞丅
丂丂丂恾俆丂擕壔塼枌朄偺憤妵摟夁梕検學悢偲暘攝斾偺娭學[10]
俇丏偍傢傝偵
C4僆儗僼傿儞偺媧廂暘棧偵偍偄偰桳岠惈偑妋擣偝傟偰偄傞嬥懏摵乛戞擇摵梟塼愙怗暘棧宯傪塼忬僆儗僼傿儞偺拪弌暘棧偵棙梡偡傞偨傔偵丄堦憌専摙傪恑傔傞昁梫偑偁傞傕偺偲峫偊傜傟傞丅偙偺曽朄偼嬥懏摵傪娷桳偡傞晄嬒堦斀墳宯偲偄偆寚揰偼偁傞偑丄楑壙偱兾嶖懱宍惉擻偑崅偄偲偄偆摵偺摿挿傪妶梡偡傞偱偒傞傕偺偲婜懸偝傟傞丅 堦曽丄斀墳惈偺崅偄摵傪梡偄傞偙偲偵傛傞晄棙塿偵偮偄偰傕専摙傪恑傔側偗傟偽側傜側偄丅偡側傢偪丄拪弌憖嶌偵傛傞僆儗僼傿儞偺曄幙偵偮偄偰挷傋丄幚嵺偵媡拪弌偑壜擻偱偁傞偙偲傕妋擣偟側偗傟偽側傜側偄丅
堷梡暥專
1) Hartley,F.H.: Chem.Reviews, 73,163(1973)
2) 壛摗丂妎,働儈僇儖僄儞僕僯傾儕儞僌,40,27(1995).
3) Eldridge,H.B., IEC.Res.,32,2208(1993)
4) Gilliland et al., J.A.C.S.,63,2088(1941)
5) 摿奐丄徍51-26806
6) 摿岞丄徍48-35041
7) 摿奐丄徍50-18425
8) 摿奐丄徍57-58633
9) Kato,S.,K.Nakano,H.Noritomi,K.Nagahama:Solv.Ext.Res.Dev.,Japan,3,117(1996).
10)Kato,S.,K.Nakano,H.Noritomi,K.Nagahama:Solv.Ext.Res.Dev.,Japan, in press.
11)Vatai,G.,M.N.Tekic:Sep.Sci.Tech.,26,1005(1991).
12)Kato,S.,J.Kawasaki,J.Chem.Eng.Japan,20,140(1987).
13)Egashira,R.H.Tanno,S.Kato,J.Kawasaki,J.Chem.Eng.Japan,28,38(1995).
丂奾漚慜偺摵暡枛乮暯嬒宎偼栺0.2mm)偼桘悈奅柺偵懡偔偑懲愊偟偰偄偨丅奾漚掆巭屻偺崌堦奅柺偺堏摦曽岦偑壓岦偒偱偁傞偙偲偐傜奾漚拞偺暘嶶偺宆偼俷乛倂宆偱偁傞偙偲偑傢偐傞丅
丂娤嶡寢壥偐傜恾俀偺傛偆側拪弌婡峔偑峫偊傜傟傞丅僆儗僼傿儞偼桘憡偐傜悈憡偵暘攝偡傞丅悈憡偺奅柺嬤朤偱嬥懏摵偲Cu(II)偑愙怗偟偰晄埨掕側Cu(I) 偑惗惉偡傞丅潣漚梼偺愭抂椞堟偱偼棳懱偺棎傟偑嫮偄偺偱Cu(I) 偺惗惉懍搙偑崅偔丄椢怓傪掓偡傞丅偙偺Cu(I) 偺堦晹偵僆儗僼傿儞偑攝埵偟偰埨掕側嶖懱偑宍惉偝傟傞丅嶖懱偼悈梟塼傪椢怓偵曄偊傞丅
恾俀丂俠倳乮侽乯乛俠倳乮俬俬乯愙怗宯偵偍偗傞僆儗僼傿儞偺拪弌婡峔
係丏俀丂拪弌懍搙[2]
恾俁偵丄侾丆俆僿僉僒僕僄儞偺拪弌棪偺宱帪曄壔偵媦傏偡奾漚懍搙偺塭嬁傪帵偡丅偙偺拪弌宯偼摵暡枛傪娷傓晄嬒堦斀墳宯偱偁傞偺偱斀墳惉暘傗嶖懱偺奼嶶偑拪弌懍搙偵嫮偔塭嬁偡傞偙偲傪帵偟偰偄傞丅偙偺揰偼Cu(0)/Cu(II)愙怗拪弌朄偺寚揰偲偟偰嫇偘傜傟傞丅恾俁丂侾丄俆僿僉僒僕僄儞偺拪弌棪偺宱帪曄壔偵媦傏偡奾漚懍搙偺塭嬁[2]
丂恾係偵侾丄俆僿僉僒僕僄儞偲侾亅僿僉僙儞偺拪弌棪偺宱帪曄壔傪斾妑偟偨丅恾係偵偼AgNO3 悈梟塼傪梡偄偨応崌偺拪弌嬋慄傕帵偟偰偁傞偑丄偙偺応崌偵偼嬒堦宯偱偁傞偐傜奼嶶偺掞峈偑彫偝偄偺偱拪弌懍搙偼嬌傔偰懍偄丅
丂恾係丂俠倳乮侽乯乛俠倳乮俬俬乯愙怗拪弌宯偲徤巁嬧悈梟塼偵傛傞僆儗僼傿儞偺拪弌棪偺宱帪曄壔[2]
係丏俁 暯峵拪弌検[2]
恾係偺Cu(0)/Cu(II)拪弌宯偺寢壥偱偼拪弌暯峵偵偼摓払偟偰偄側偄偑丄廫暘偵愙怗偣傞偲俙倗乮俬乯嶖懱偵斾傋偰暯峵拪弌検偑崅偔側傞孹岦傪帵偟偰偄傞丅偙偺寢壥偼俙倗乮俬乯偵斾傋偰俠倳乮俬乯偺曽偑嶖宍惉掕悢偼戝偒偄偲偄偆堦斒揑孹岦[1]偵崌抳偟偰偄傞丅丂恾係傪傒傞偲丄俠倳乮俬乯偵偍偄偰傕俙倗乮俬乯偲摨條偵丄侾丄俆僿僉僒僕僄儞偺曽偑侾亅僿僉僙儞傛傝暯峵拪弌検偼挊偟偔戝偒偄偺偱丄俠倳乮俬乯亅侾丄俆僿僉僒僕僄儞偺僉儗乕僩傪宍惉偟偰偄傞壜擻惈偑崅偄丅幚嵺偵丄俠倳乮俬乯偲侾丆俆僿僉僒僕僄儞偺寢崌斾棪偼侾丗侾偱偁傞偙偲偑柧傜偐偵偝傟偰偄傞[9]丅僩儖僄儞側偳懠偺惉暘偵偮偄偰偼暘愅惛搙撪偱拪弌偝傟偨挍岓偼尒傜傟側偐偭偨丅
俆丏擕壔塼枌暘棧朄傊偺墳梡
丂俠倳乮俬乯僀僆儞傪娷傓倧乛倵乛倧擕壔塼枌朄偵傛偭偰侾丆俆亅僿僉僒僕僄儞傪暘棧偡傞偲憐掕偟偰丄婛懚偺擕壔塼枌摟夁僨乕僞偵擣傔傜傟傞娭學偐傜拪弌懍搙傪悇掕偟偰傒傞丅丂Kato傜[10]偼丄婛墲偺曬崘偵偁傞擕壔塼枌摟夁僨乕僞傪梡偄偰憤妵摟夁梕検學悢俲i倎傪媮傔丄奼嶶棩懍偺忬懺偱偁傟偽俲i倎偲暘攝斾俢i偺娫偵師幃偺憡娭娭學偑惉傝棫偮偙偲傪尒偄偩偟偰偄傞丅
偙偺娭學傪恾俆偵帵偡丅堦曽丄Cu(0)/1M-Cu(NO3)2宯偵傛偭偰侾丄俆僿僉僒僕僄儞傪拪弌偡傞嵺偺暘攝斾偼侽丏俁 掱搙偱偁傞偐傜[9]丄恾俆忋偱乮侾乯幃傪枮偨偡擕壔塼枌摟夁僨乕僞偲斾傋傞偲丄俢俀俤俫俹俙側偳偵傛傞媯傒忋偘岠壥傪棙梡偡傞傾儈僲巁偲摨偠掱搙偺摟夁懍搙傪帵偡偲梊憐偝傟傞丅偨偩偟丄廫暘偵斀墳懍搙傪崅傔偰嬒堦斀墳宯偵偍偗傞奼嶶棩懍忬懺偑幚尰偝傟傞偲壖掕偟偰偄傞丅側偍丄乮侾乯幃偺娭學偼暘嶶偺宆(w/o/w,o/w/o)偵埶懚偣偢丄崅偄摟夁懍搙傪幚尰偡傞憖嶌忦審偱偁偭偰奼嶶棩懍忬懺偵偁傟偽亇俆侽亾偺惛搙撪偱擣傔傜傟傞.傑偨丄拞嬻巺儌僕儏乕儖[11]偵斾傋偰傕擕壔塼枌偺梕検學悢偼崅偔丄僄儅儖僔儑儞揌宎乛撪晹塼揌宎偺斾傪侾偵嬤偯偗傞偙偲偵傛偭偰暘棧學悢傪偨偐傔傜傟傞[12,13]偙偲偑帵偝傟偰偄傞丅
丂丂丂恾俆丂擕壔塼枌朄偺憤妵摟夁梕検學悢偲暘攝斾偺娭學[10]