「化学教育ジャーナル(CEJ)」第2巻第1号/採録番号2-10/1998年6月30日受理
URL = http://www.juen.ac.jp/scien/cssj/cejrnl.html
ネットワークソフトウェアの化学教育における有効利用に関する考察
立花 宏
(東京都立大学大学院工学研究科分子変換化学大講座)
ソフトウェアは、ある程度使い込んではじめて良さ悪さがわかり、ハードウェアのようにスペックでは判断はできない面があります。しかし一般的に研究教育予算はハードウェアが中心であってソフトウェアに重点を置きにくくなっています1)。一旦納入されたシステムは、ハードとソフトのどちらかに問題があってもうまく機能せず、解決はなかなか困難です。唯一の解決手段は、物を必要としないソフトウェア面での工夫と考えられます。 必要なソフトウェアは、自分で1からプログラミングして作るのが理想ではありますが、なかなか大変な作業であるため事実上不可能です。そのため従来は、宣伝されているものの中から適当なものを購入するしか方法がありませんでした。現在はインターネットが普及して、個人レベルでの広範な情報収集が可能になってきましたので、ソフトウェアに関する情報も以前に比べ格段に多く得られるようになりました。その中には、研究者などが個人で作ったソフトウェアを無料で配布している、または、メーカーが販売促進のために宣伝として製品の一部分を無料で配布しているものがかなりあります。有料であっても、流通宣伝経費がかからないので非常に安い価格設定がされています。これらは、統合環境を持った高価なソフトウェアに比べると、機能の面では遜色なくとも使い勝手やマニュアルの不備(外国語ということも含む)の面で劣る場合が多いという欠点があります。しかしこの欠点は、インターネットのWWWなどの機能を使った多くの利用者による協調と分業作業で補完できるものです。
ネットワーク上には様々な化学関連情報があります2)3)4)。その中には、本格的データベースとしては最も歴史のある部類に属するケミカルアブストラクトデータベース(CAS)を始めとする有償の情報、そして、閉じたネットワークであるパソコン通信上での化学・化学教育関係の情報交換やソフトウェアの流通があります。これらに関しては広範で自由な利用ができませんので本稿では扱っていません。
化学の教育では、分子に対する親しみと感覚を持てるようになることが重要です。このためには分子模型キットが有用なので販売もされています。しかし、どのような分子でも作れるわけではなく、1つのキットでは作れる模型の種類もボール&スティック型等に限られてしまうため、汎用性に欠け、各人が購入するほどの魅力はないものと思われます。パソコン上の分子模型ならば、実際に手に持っていじるような感覚は持てないものの、拒絶感は少ないと考えられます。他にもパソコンを使っていれば、レポート作成時に分子式、構造式やグラフを綺麗に作成することができる点などを考えると、卒業研究以前の学部生や高校生などが早いうちに化学用ソフトウェアを体験しておく事は意味のあることだと考えられます。 パソコンの普及率が上がってきている今、ソフトウェアに関する情報が行きわたれば可能なことです。
WWWを使うとだれもが化学情報を集められ、提供する側にもなれます。この現在のインターネットの発展の元になったWorld-Wide Webシステムは、1993年に発表されたものですが、その時点で既に、画像、動画や音声も扱えるハイパーリンクが実現していました。一方、電子メールでは文字情報での通信しか行えませんが、文字以外の情報を文字化して多種多様の情報をやりとりする仕組みであるMIME(Multipurpose Internet Mail Extensions)が同じ年に考案されました。この機能は、直ちにWWWにも取り入れられました。そして翌年の1994年には、MIMEを化学の分野に生かすためにChemical MIMEが提唱され、多くの化学に関する情報をWWWで扱う準備ができあがったわけです。まだ一般にはインターネットが知られていない時代から利点を見抜いて、化学に特化した利用のための実験、研究が進められていました。このChemical MIMEとは、分子の座標や結合、分子のモデル化、スペクトルなどを扱う形式を定めたものです。MIME typeには、chemicalが定義されています。その中の分類にはsubtypeと呼ばれ、x-pdb (Protein Database 形式), x-mdl-molfile (MDL Molのファイル形式)など多くの化学関係のアプリケーションやデータタイプがあります。また、chemical以外のMIMEタイプで化学に利用されているものには、VRML(汎用の仮想空間設計言語)のmodel/vrml、x-world/x-vrml、JAVAのapplication/x-javascriptなどがあります。自分のブライザがどのChemical MIMEタイプを扱えるかを知りたい場合には、Chemical Object Test Pageで確認することが出来ます。また、ブラウザやサーバのMIME設定が正しく行われていない場合には、正しく表示されない場合があります。
このChemical MIMEを使って3次元分子模型を転送する仕組みが考案されています。これらの手法の利点は3つあります。第1に、ファイルサイズが小さいのでネットワークを介したデータ転送に有利であること。原子数数百までの場合は、画像ファイルよりもサイズが小さくできます。第2に、静止した画像でなく、画面上で動かすことによって分子構造をより細かく理解できる点。第3に、座標データを他のプログラムなどで再利用することができる点です。これからもこの分野は、ネットワーク技術と共に急速に進歩することでしょう。
ホームページ上での3次元分子模型
次に、ホームページ上での3次元分子模型について説明します。
以後、Macintoshを例にしましすが、かなりのソフトウェアはWindows版またはUNIX版もありますので同様の方法で利用することができます。特に断らない場合は全て無料ソフトウェアですが、使用条件など詳しくは各ソフトウェアの添付文書を参照下さい。
ホームページ上で動かせる3次元分子模型を表示させたり、作る方法は、大まかに分類して次の3種類があります。1)ブラウザ側で用意したプラグインソフトを使用する方法、2)JAVAを使う方法、3)VRML(Virtual Reality Modeling Language)を利用する方法。
- )
 ブラウザのプラグインソフトであるMDL社の Chemscape Chimeを使う方法(PC版もあり)
ブラウザのプラグインソフトであるMDL社の Chemscape Chimeを使う方法(PC版もあり)
MDL Chemscape ChimeをブラウザNetscapeやInternet Explorerにインストールすることによって数々のファイル形式の分子座標ファイルをブラウザ上で模型として見ることができるようになります。Chemscape Chimeは、http://www.mdli.com/tech/chemscape.htmlにあり、ダウンロードページは、ここです。インストール以降、ブラウザが下記のようなHTML行を読み込むと自動的に3次元分子模型を表示するようになります。自分のホームページ上に作る場合も同様です。
<EMBED SRC="test.mol" display3d= ball&stick width=100 height=100>
これらのパラメータの意味は、
分子座標ファイル名 test.mol
表示方式 ball&stick
幅 300dot
高さ 300dot

図1 Chemscape Chimeによる3次元分子模型の例
分子模型の表示されている矩形の中をマウスをクリックした状態で動かすと、分子模型を自由に回転させることができます。操作法は、マウスをクリックした状態で動かすと自由回転、オプションキー+マウスクリックで平行移動、シフトキー+マウスクリックで拡大縮小、シフトキー+オプションキー+マウスクリックで画面平面内回転です。
また、マウスを動かさずにクリックし続けると、メニュー画面が現れて、座標ファイルを保存したり(図3)、表示形式を変えたり(図4)、ファンデルワールス半径の位置を点表示する(図5)ようなこともできます。
図3 ブラウザに表示されている分子模型から
座標データを保存しているところ
Chemscape Chimeを使ったホームページは数多く見られ、化学の研究教育等に活用されています。5)6)7)
メニュー画面に表示されるOptionやRotationを書き加えておくことによって最初に表示される模型を変化させることができます。分子座標は、MOL形式、XYZ形式など多種類が使えます。分子座標ファイルを作る方法については後述します。Chimeが使える分子座標形式の一覧は、ここにあります。
- )
 JAVAを使った分子模型を見る場合にはJAVAに対応したブラウザ(より新しいバージョンの方が安定した動作をします)を使うことによって、見る側はなにも準備する必要はありません。ホームページの表示に使う場合には、Cherwell Scientific社のChemsymphonyの利用が便利です。(PC、UNIX上でも動作する)JAVA言語の知識は必要ありません。無料公開されているLite版を取得後、解凍するとRenderBasic.classを始めとして多数のJAVAアプレットとGIFファイルが現れます。この中のstart_here.htmlをブラウザで読み込むことによって利用法を知ることができます。
JAVAを使った分子模型を見る場合にはJAVAに対応したブラウザ(より新しいバージョンの方が安定した動作をします)を使うことによって、見る側はなにも準備する必要はありません。ホームページの表示に使う場合には、Cherwell Scientific社のChemsymphonyの利用が便利です。(PC、UNIX上でも動作する)JAVA言語の知識は必要ありません。無料公開されているLite版を取得後、解凍するとRenderBasic.classを始めとして多数のJAVAアプレットとGIFファイルが現れます。この中のstart_here.htmlをブラウザで読み込むことによって利用法を知ることができます。
ホームページに分子座標データtest.molを表示させるには下記のようにHTML言語を記述します。
<APPLET CODE=RenderBasic.class WIDTH = 300>
<PARAM NAME=model value=test.mol>
</APPLET>
1行目のRenderBasic.classから読み込まれた後、インストールされている他の多数のJAVAアプレットが自動的に読み込まれます。これらのファイルはHTMLファイルと同じディレクトリ階層にある必要があります。多数のファイルを置いておくのは煩雑なので、代わりにこれらのファイルをまとめてあるchemsymphony.lite.zip ファイルだけを置いておくこともできます。
その場合は、オプションパラメータとしてarchive=chemsymphony.lite.zipと指定します。
<APPLET CODE=Renderbasic.class ARCHIVE=chemsymphony.lite.zip WIDTH=300>
このzipファイルはMacintoshサーバー上でも動作しました。
2行目の value= で分子座標ファイルを指定します。
画面上での分子模型の操作法は、
マウスクリックで自由回転
シフトキー+マウスクリックで拡大縮小
コントロールキー+マウスクリックで移動です。
図6 JAVA(ChemsymphonyLite)による分子模型表示例
現在のネットワークの回線速度では、アプレットの転送に時間がかかり、応答が少し遅くなりますが、分子模型を見る側からすれば、プラグインソフトのインストールなどの準備が必要ないため有利です。
なお、JAVAの本家であるSunの3D Molecule Viewerを使って分子模型を表示することもできます。
- )VRMLを使った表示
VRMLには現在VRML1.0仕様とVRML2.0仕様があります。1.0は、静止3次元モデルをマウスで動かすことのできるもので、2.0では、マウスの動きに対してインタラクティブな反応をさせたり音を扱うことができます。
ブラウザーのプラグインソフトには数種類あり、NetscapeNavigator付属のLive3Dや、ExpressVRではVRML1.0を、WorldViewでは、VRML2.0を使用することが出来ます。
VRML用の分子座標ファイルを作るには、後述のWebLab Viewerで読み込んだファイルをVRML形式で出力することでVRML1.0ファイルの作成ができます。
また、数少ない国産のフリーの3次元分子模型作成ソフトMOLDAが、JAVAによって書き直され、Windows版のみでなくMOLDA Beansとしてプラットフォームを選ばなくなりました。Macintosh版のMOLDA on Macは、http://cssj.chem.sci.hiroshima-u.ac.jp/MoldaBeans/mac-j/にあり、使用法、応用例等の解説があり、VRML形式のファイル出力が行えます。PDB、Xmolなどのファイル形式にも対応しています。
PDB形式のファイルとはBrookhaven National Laboratoryで作られているProtein Data Bankで使用されているファイル形式で、広く用いられています。
分子の構造式、3次元模型データの作成と表示
 まず、2次元の化学構造式を書くために、ISIS/DrawをMDL社のサイトから取得します。
まず、2次元の化学構造式を書くために、ISIS/DrawをMDL社のサイトから取得します。
このソフトはChemDrawとほぼ同等の機能を持っており2次元の構造式を書く機能に優れています。前述のMOLDAでは直接3次元構造を作れますが、2次元構造式を作る作業の方が手書きに近く、はじめて使う学生にとって抵抗が少ないだろう点、また、文章の挿入や画像の張り込みが可能なので簡単なレポート作成にも役立てられる点を考えISIS/Drawを入力用ツールとして選んでいます。
ISIS/Drawは、3D-Rotate Toolによって選択部分を3次元回転させることができるので、使い勝手に多少の難はありますが立体的な分子構造を書くことができます。
図7 C6H12の入力例
図8 C6H12の3次元回転ツール
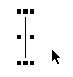
図9 イス型C6H12の作成法
分子構造ファイルを出力するには、出力したい分子を選択した状態で、Export機能を使ってMOL形式の分子座標ファイルに出力します。このとき、目的の分子を選択していないとExportメニューの中でMOL形式が選択できませんので注意が必要です。
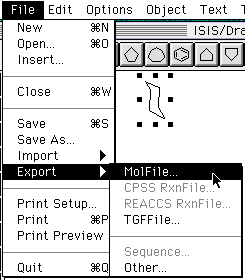
図10 MOL形式での出力方法
 また、ISIS/Drawと共にRasMacという3次元分子模型表示ソフトが同時にインストールされるので、これを使って2次元構造式を動く3次元模型とすることもできます。
また、ISIS/Drawと共にRasMacという3次元分子模型表示ソフトが同時にインストールされるので、これを使って2次元構造式を動く3次元模型とすることもできます。
 ここでは、WebLab Viewerを使ってみます。
ここでは、WebLab Viewerを使ってみます。
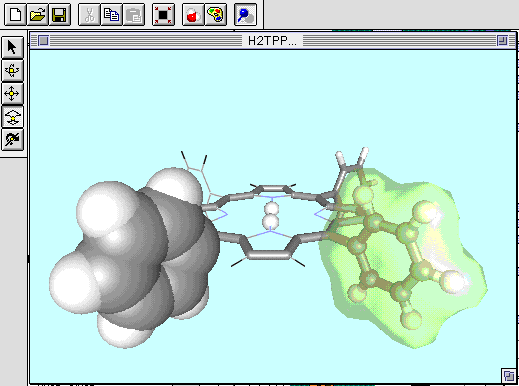
このソフトは、市販ソフトでも持っていないものが多い1つの分子中での表示方法を原子毎に変える機能があるので、一部分を強調したり、ワイヤーフレーム表示で目立たなくするなどの表現が可能です。
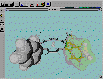
図11 WebLabViewerによる分子模型例
また、PDB形式やMOL形式ファイルも読み込むことができ、ISIS/Drawから直接構造をCopy&Pasteすることもできます。
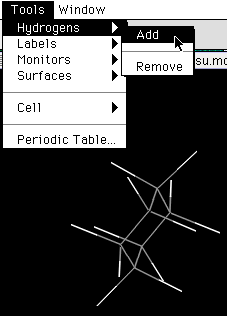
ISIS/Drawで作ったMOLデータには、水素原子が含まれていませんが、WebLab ViewerのメニューのTools->Hydrogens->Addで水素原子を自動的に付加することが出来ます。
図12 WebLab Viewerでの水素原子の自動付加
(さらにその後、WebLabViewer上でMOL形式で保存して、ISIS/Drawで読み込み(import)し直せば水素原子を含んだ構造をISIS上でも使うことができます。)
そして、WebLab Viewer上で、MOL形式でのファイル出力を行います。このソフトは、他にPDB、VRML、GIF、JPEG(この2つはWWWで標準的に使われている画像ファイルなのでそのままホームページの静止画像に使用できる)などのファイル形式で出力ができます。
作ったファイル(temp.mol)を自分のホームページのあるディレクトリにもって行き、下記のようなHTMLを付け加えれば3次元分子模型のあるホームページが完成します。
< EMBED SRC="tmp.mol" display3d= ball&stick width=300 height=300>
他にも3次元表示を行えるソフトウェアがインターネット上で入手できます。上述のものも含めて列記し、画面上での分子模型の動かし方を併記しています。
下記の3次元分子模型表示ソフトは、いずれも蛋白、DNAなどの表示を考えて作られているので小さい有機分子の描画はSpacefillモードでも非常に高速です。
- WebLab Viewer
上述
マウス操作は、画面上のボタンにより選択する。
- Rasmac(PC版もあり)
PDBとMOL形式で入出力が出来る。
マウスをクリック状態で 自由回転
Optionキーで移動
Optionシフトで画面上の平面を回転
シフトクリックでズーム
スクロールバーで軸方向の回転
 Swiss-PdbViewer(PC版もあり)
Swiss-PdbViewer(PC版もあり)
PDBファイルを読み込み、
原子間距離や角度をマウスクリックで表示できる。
出力形式POV3で出力したpovとincファイルは、POV-Rayでレンダリングに使える。
画面上のボタンによってマウスの機能を変更する
 MacMolecule2 表示例(図13)
MacMolecule2 表示例(図13)
MacMolecule2の機能縮小版です。
PDBファイルを読み込んで表示することができます。
マウスをクリックした状態で自由回転
シフトクリックでz軸回転
Optionキークリックでx軸回転
 MicroWorld
MicroWorld
DEMO版では80原子まで、登録すると2500原子まで扱えます。PDB形式ファイルを読み込め、3DMF形式ファイルに出力できます。3DMF形式は、3Dモデリングの分野で広く用いられているファイル形式でMacOSに標準添付されているQuickDraw3DがインストールされていればSimpleTextなどのエディタ上でも3次元表示(回転)ができます。
シフトクリックで画面回転
Optionで移動
Optionクリックでズーム
 ChemViewer3D 表示例(図14)
ChemViewer3D 表示例(図14)
$50のシェアウェアで登録しないと保存、印刷ができない。MicroWorldユーザーは$40で入手可
PDBデータなどを読み込んで3次元模型表示を行うソフト。
原子ごとに表示方法を変更したり(ワイヤーフレームとスペースフィルを組み合わせるなど)、光源の方向をコントロールキー+マウスで簡単に変えられるなど、多少凝った分子画像を手軽に作れる。
シフトで自由回転
シフトクリックで画面平面内回転
Optionキーで移動
Optionクリックでズーム
(登録後3DMF形式のファイル出力ができる)
 MAGE(PC,UNIX版もあり)
MAGE(PC,UNIX版もあり)
蛋白質用。PDBファイルを付属のPrekinプログラムでKinフォーマットに変換して読み込める。
これらのソフトは、上記のように同じ3次元模型を動かす操作だけで見ても各々が異なっています。動かすだけの操作ではそれ程の抵抗はないですが、もっと複雑な操作を必要とする立体分子構造を作る機能を持った市販のChem3D、CAChe、MacSPARTAN、WinMOPAC、CERIUSなどでも全てが異なっています。そのため、一つのソフトを使いこなしていたとしても他を使いこなすまでにはかなりの習熟を要するということになりソフトウェア利用の敷居を上げています。わかりやすく便利な操作性を持つことは、利便性のために作られているソフトウェアにとって重要な問題です。
分子の最適化構造の作成
上記で作った分子模型の構造は適当に作成したもので、正確ではありません。そこで、最適化構造を求める計算を行う必要があります。数々の方法があり、その計算法の精度に応じた構造を得ることができます。計算時間と必要メモリー量の少ない順にあげると、分子力学計算、半経験的分子軌道計算、非経験的分子軌道計算があります。分子力場計算プログラムにはTINKERがあり、Macintosh上でも動作しますが、入力ファイルの作成が多少面倒になるので割愛します。量子化学計算を行える無料配布されていて比較的簡単に使えるプログラムには、半経験的量子化学計算プログラムのMOPAC Ver6, Ver7、非経験的量子化学計算プログラムのGAMESS(MacGAMESS)などがあります。
MOPACは、だれにでも接続できるAnonymous Ftpサーバ上にあるので下記をブラウザに入力して取得します。

 FTP to 東北大学またはMacintosh The Chemical Tune-Up
FTP to 東北大学またはMacintosh The Chemical Tune-Up
入力ファイルとしては、簡単のため水分子を例として説明します。
まず、ISIS/Drawを使って酸素原子Oだけを入力します。
図15 ISIS/Drawで酸素原子を入力する
これをExportメニューでMOL形式ファイルとして書き出します。このファイルをWebLab ViewerにDrag&Dropして読み込ませ、メニューのTools->Hydrogens->Addで水素原子を付加してからPDB形式でファイル出力します。その後MacBabelを使ってMOPAC Internal形式に変換します。(なお水素原子の付加はMacBabelでもできますが、WebLab Viewerではモデル上での確認ができます)先ほどの手順で作ったPDB形式の分子座標ファイルをMacBabelでMOPAC Internal形式に出力したものが下記です。(ファイル名はH2O.inp)
 MacBabelは、多くのファイル形式間での変換を行うソフトです。汎用に使えるので常備しておくと便利です。(PC、UNIX版もあり、さらに変換できるファイルの種類が多いようです)
MacBabelは、多くのファイル形式間での変換を行うソフトです。汎用に使えるので常備しておくと便利です。(PC、UNIX版もあり、さらに変換できるファイルの種類が多いようです)
図16 MacBabelで読み込めるファイル形式
図17 MacBabelで書き出せるファイル形式
-----次の行から-----
(この行は空行)
Macintosh HD:MacBabel no FPU 1.03 ト:H2O.inp
Macintosh HD:MacBabel no FPU 1.03 ト:H2O.inp
O 0.0000000 0 0.000000 0 0.000000 0 0 0 0 0.0000
H 0.9900020 1 0.000000 0 0.000000 0 1 0 0 0.0000
H 0.9899753 1 109.532960 1 0.000000 0 1 2 0 0.0000
-----ここまで-----
MOPACでは1行目に計算方法などのキーワードを与える必要があります。
ここでは、適当なエディタでブランクになっている1行目に
EF AM1
を加ます。これらの意味は、EF : 固有ベクトル追跡法で安定構造を求める、AM1のハミルトニアンを使用するということです。なお、遷移金属などの計算できない原子があります。詳しくは、分子軌道法MOPACガイドブック 平野恒夫・田辺和俊編 海文堂 を参照下さい。
次にmopacを起動して下記のように入力ファイル名をキーボードから入力してリターンキーを押すと下記のように計算が行われます。(Drag&Dropでは計算は始まりません)
-----次の行から-----
INPUT FILENAME
H2O.inp
EF WORKING....
CYCLE: 1 TIME: 0.00 TIME LEFT: 7200.0 GRAD.: 46.804 HEAT:-57.51358
CYCLE: 2 TIME: 0.00 TIME LEFT: 7200.0 GRAD.: 29.690 HEAT:-58.37735
CYCLE: 3 TIME: 0.00 TIME LEFT: 7200.0 GRAD.: 10.122 HEAT:-59.06605
CYCLE: 4 TIME: 0.00 TIME LEFT: 7200.0 GRAD.: 7.764 HEAT:-59.11954
CYCLE: 5 TIME: 0.00 TIME LEFT: 7200.0 GRAD.: 0.278 HEAT:-59.24074
COMPUTATION TIME = 0.000 SECONDS
== MOPAC DONE ==
-----ここまで-----
一瞬で計算は終わり、詳細な計算結果がファイルFOR006に、主要な結果がファイルFOR012に出力されます。FOR012をSimpleTextなどのエディタで開いて後半部分の入力ファイルと同じ形式になっているEF AM1という行以降だけを残して前半部分を削除します。
-----次の行から-----
EF AM1
Macintosh HD:MacBabel no FPU 1.03 ト:H2O.inp
Macintosh HD:MacBabel no FPU 1.03 ト:H2O.inp
O 0.0000000 0 0.000000 0 0.000000 0 0 0 0 -0.3826
H 0.9614617 1 0.000000 0 0.000000 0 1 0 0 0.1913
H 0.9614647 1 103.504252 1 0.000000 0 1 2 0 0.1913
-----ここまで-----
このファイルをMacBabelを使ってPDB形式に変換すれば、構造最適化された3次元模型をWebLab Viewer、MacMoleculeなどのプログラムで自由に模型として動かすことが出来るようになります。
この場合はファイル内の数値を見るとHOHの結合角は103.5度と自明ですが、複雑な分子の場合にはSwiss-PdbViewerを使って結合角、原子間距離などを測ることもできます。
 MacGAMESSは、非経験的量子化学計算ソフトGAMESSのMacintosh版です。(PC、UNIX版もあり)
MacGAMESSは、非経験的量子化学計算ソフトGAMESSのMacintosh版です。(PC、UNIX版もあり)
ハミルトニアンには、RHF, ROHF, UHF, GVB, MCSCF, CI, MP2、
基底関数には、STO-3G, 3-21G, 6-31G*, 6-311G(d), DZV, DH, BC, TZV, MCなどが使え、これらのパラメータの選び方によって半経験的方法よりも精密な計算ができます。
上記からGAMESSのアーカイブファイルは取得できますが、ファイルを解凍するにはパスワードが必要です。
パスワードは、開発者のMike Schmidt氏宛(mike@si.fi.ameslab.gov)にE-mailで請求できます。使用に当たっては、直筆のサイン入りの使用許諾書を郵便で送る必要があります。
入力ファイルの形式は、MOPAC形式が使えるので前述のMOPACでの計算結果を再利用します。
ファイル名をH2Otest.inpとします。
-----次の行から-----
$CONTRL SCFTYP=RHF RUNTYP=OPTIMIZE UNITS=ANGS COORD=ZMTMPC $END
$SYSTEM TIMLIM=600 MEMORY=200000 $END
$SCF DIRSCF=.True. $END
$GUESS GUESS=HUCKEL $END
$BASIS GBASIS=STO NGAUSS=3 $END
$DATA
H2Otest
C1
O 0.0000000 0 0.000000 0 0.000000 0 0 0 0
H 0.9614617 1 0.000000 0 0.000000 0 1 0 0
H 0.9614647 1 103.504252 1 0.000000 0 1 2 0
$END
-----ここまで-----
各行の$の前にはスペースを一つ入れて下さい。
$CONTRL行の意味は、制限付HartreeFock計算、構造最適化を行う、単位はオングストローム、座標はMopacのZ-matrix
$SYSTEM行では制限時間と使用メモリーを指定
$SCF行ではSCFの種類
$GUESS行では初期値の決め方
$BASIS 基底関数はSTO型でガウス関数を3つ使う(STO-3G)
$DATA行以降にファイル名、分子の対称性(水分子の対称性はC2vですが、入力時に対称性を考える手間を省くため全原子を入力して対称性をC1にしてあります。)
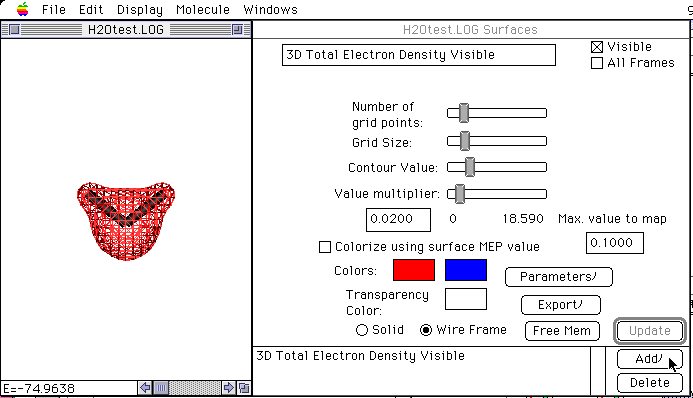
計算結果は、ファイルH2Otest.LOGに出力されます。
 この計算結果ファイルを、専用の可視化ソフトMacMolPlt読み込ませることによっていろいろな3次元表示をすることが出来ます。
この計算結果ファイルを、専用の可視化ソフトMacMolPlt読み込ませることによっていろいろな3次元表示をすることが出来ます。
分子の3次元構造のみならず、電子密度や、分子軌道の3次元表示もできます。
そのためには、AppleのQuickDraw3Dがインストールされている必要があります。(MacOS CD-ROMに入っています)
まず、計算結果の.LOGファイルをMacMolPltにDrag&Dropします。
メニューからDisplay->Use QuickDraw 3Dを選んだ後、メニューのWindows -> H2Otest.LOG -> Surfacesを選び、Window中のAddボタンをクリックして表示したいSurface typeを選びUpdateボタンをクリックする。
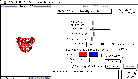
図18 水分子の全電子密度表示例
3次元モデルは、メニューからExportを選んで保存することによって電子密度表示も含めた形で3DMFファイルに保存できます。この3DMFファイルは、QuickDraw3DがインストールされていればSimpleText等でも同様に動かせる3次元モデルとして利用できます。
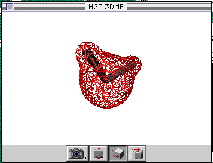
図19 SimpleTextによる3DMFファイル化した水の分子軌道
3次元座標を他の表示ソフトで利用するには、File->Export でXMol .xyzタイプで出力する。この時、All Framesのチェックは外しておきます。
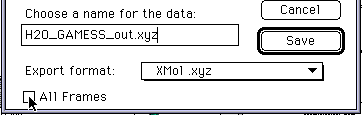
図20 MacMolPltを使ってMacGAMESSで構造最適化した
座標をxyz形式ファイルに出力する
次に、MacBabelを使ってXYZ形式で読み込み、PDB形式等で出力することによって汎用の座標ファイルとなります。
高品位静止画像の作成
量子化学計算によって分子構造が最適化されました。この構造を希望の方向から眺めた高品質な静止画像にします。手順としては、まずモデリングプログラムによって表面の座標データや光源との関係を計算してモデル情報を作った後、レンダリングプログラムによって明るさや色を持ったテクスチャーを作り出すという2段階の操作になります。分子座標ファイルにはPDB形式を用います。モデリングはMacSceneまたは、Swiss-PDBViewerまたはpdb2pov(動作未確認)で行うことができます。
 MacSceneでは、PDBファイルを読み込み後、表示方式、方向や光源、背景色などを決めることができます。
MacSceneでは、PDBファイルを読み込み後、表示方式、方向や光源、背景色などを決めることができます。
表示方向を決定するPreview画面の操作法は下記のとおりです。
マウスクリックで自由回転
シフトキー+マウスクリックでズーム
Optionキー+マウスクリックで移動
シフトキー+Optionキー+マウスクリックで、画面平面内回転
Save Fileボタンをクリックすると2つの出力ファイルが生成されます。
例えば、Untitled.pov、Untitled.inc
次にこれらを汎用のレンダリングソフトPOV-Rayを使って美しい画像にします。
 POV-Rayの正規版はhttp://www.povray.org/にありますが、ソースコードも公開されているので、色々な改良が施されたものが作られています。ここでは、PovMac Unofficialの方が高速で使いやすいのでこちらを使います。
このUnofficial版は、正規版のscenes, includes, official docsファイルを使用しているので両方をもらってくる必要があります。正規版は、Unofficial版のホームページからリンクされているhttp://ourworld.compuserve.com/homepages/povraymac/からもらってくるのがいいでしょう。手順は以下の通りです。
POV-Rayの正規版はhttp://www.povray.org/にありますが、ソースコードも公開されているので、色々な改良が施されたものが作られています。ここでは、PovMac Unofficialの方が高速で使いやすいのでこちらを使います。
このUnofficial版は、正規版のscenes, includes, official docsファイルを使用しているので両方をもらってくる必要があります。正規版は、Unofficial版のホームページからリンクされているhttp://ourworld.compuserve.com/homepages/povraymac/からもらってくるのがいいでしょう。手順は以下の通りです。
- ドキュメントセットと実行ファイルを自分のMacの種類に合わせてダウンロードする。
- ドキュメントファイルを解凍した中のIncludeフォルダの"中身"を実行ファイルPovRay PPC Unofficialと同じフォルダに入れる。
- 先ほど作った*.povと*.incファイルを同じフォルダに持ってきておく。
- PovRay PPC Unofficialを起動する。
- メニューの File->Rendering Preferences で画像のサイズを指定する。(400 X 400 など)
- Use Anti-aliasing(smoothing)をチェックすると演算時間がかかるがよりなめらかになる。
- FIle->Files and Pathsの下から3番目の項目のInputに先ほど作った*.povファイルを指定する。
- FIle->Files and Pathsの上から2番目の項目のInclude Pathが設定されていなかったらincludesフォルダの場所を設定する。
- File->Raytrace で描画を開始します。(Includeファイルなどが正常な場所にないときはエラーメッセージが表示されます)
画像が作成されたならば File->Save Image Window(PICT) で保存することによって、ワープロ等他のアプリケーションで利用できます。


図21,22 作成した画像例
前述のようにSwiss-PdbViewerでも同様にpovファイルとincファイルを作成できるのでPovMac Unofficialでレンダリングを行えます。
他に、pdb2povというPDBファイルをpovファイルに直接変換するプログラムもあります。
その他の有用なソフトウェア
その他にも有用な無料は無数にありますがその中から主な物として下記のようなソフトウェアがあります。
URLの明記していない物は、Macintosh用ソフトウェアを集めてあるInfo-Macアーカイブを通して流通しています。Info-Macアーカイブは、世界中にミラーサイト(同一内容のサイト)があり、あらゆる種類のソフトウェアが登録されています。
例
理化学研究所のソフトウェアアーカイブリスト
理化学研究所にあるInfo-Macアーカイブの科学ソフト
InfoWebにあるInfo-Macアーカイブの科学ソフト
MITにあるInfo-Macアーカイブ
その他に化学関係のプログラムの交換を目的として作られたQCPE 量子化学プログラム交換機構、JCPE -日本化学プログラム交換機構でも数々のプログラムを見つけることができます。
- TINKER Molecular Modeling Package
分子力学法で構造最適化、分子動力学計算が可能。MM2、MM3などのパラメータファイルが付属しているが、パラメータのない結合を含む分子(例えばアニリン)の計算はできません。数種類のプログラムの中から収束手法を選べます。(PC、UNIX版もある。UNIX版のBabelを使うとTINKER形式の入力ファイルを作ることが出来る)
http://dasher.wustl.edu/tinker/
- ChemLetter
テキストだけで化学構造式を書くプログラム(E-mailなどで構造式を使いたい場合に利用できます)
- FoldIt (light)
蛋白質のモデリング用プログラム
- HMO-plus
経験的分子軌道計算プログラム Huckel法
統合環境上で分子の作成から結果の表示までができます。
- Mac YAeHMOP
経験的分子軌道計算プログラム 拡張Huckel法
http://overlap.chem.cornell.edu:8080/yaehmop.html
Babelなどで作ったGaussian形式のZ-matrixファイルを入力ファイルとして利用できます。
- Gaussian Companion
非経験的分子軌道計算プログラムGaussianの振動解析ファイルからIRスペクトルなどを表示します
- MOLCAT PowerMac、MAKENEW、PSI-888)
MOPACの出力ファイルから軌道図などを表示
- MCF
実験データなどのカーブフィッティングを行うプログラム
- Protractor
ディスプレイ上に表示する分度器です。画面上で分度器を使って結合角を測ることなども可能
- SwaN-MR
NMRシミュレーションプログラム
- NIH Image
有名な汎用の画像処理プログラム.(モノクロ画像用)
http://rsb.info.nih.gov/nih-image/
医療や生物関係の分野でよく使われていますが、画像中の粒子数のカウントの他、薄層クロマトグラフィーのスポット分析に使う試みもあります
- mc (Matrix Calculator)
http://www.akita-noken.go.jp/provide/mc/
秋田県立脳血管研究センターで開発されたデータ処理言語で、ソフトウェアで関数の作図から2,3次元グラフ作成、行列演算、統計計算、微分方程式の数値解法、非線形最小自乗法、画像解析、動画の作成など非常に多機能です。
- Chipmunk Basic
BASIC言語.行番号が必要であるが、グラフィック機能も一通りあるのでN88BASICからの移植も可能である。
- Msh
UNIXのShellをMac上で実現しています。tail, grep、リダイレクト、パイプなどのコマンドが使えるので、量子化学計算の出力ファイルなどの膨大なテキストファイルから必要部分を切り出すのに使うことができます。
- Atom in a Box
水素原子の軌道をs軌道からg軌道まで3次元描画、位相によるカラー表示、回転、2色の色めがねで立体視もできます。スライス断面の表示、スペクトル表示などの機能もあります。
図23 5f軌道の表示例
他に化学情報をネットワーク上で扱う取り組みとして英国のThe Open Molecule Foundationでは、CML(Chemical Markup Language)が考案されており、ロンドン大学ではCINEMAという蛋白やDNAの表示作成ソフト(UNIXサーバー上で動き、ユーザーはブラウザからJAVA Appletで操作するためプラットフォームを選ばない)が作られているなど、世界中でインターネットの機能を化学に活用する試みがなされています。
このように化学の研究・教育に活用できる有用な無料ソフトウェアは多岐にわたりって多くの数が、ボランティアの手や企業の製品戦略によって作られています。全てを理解して使いこなすのは不可能ですが、ある1つの目的を実現するために必要なものは2~3もあれば充分です。しかし、無料ソフトウェアを活用できるからといって、購入時のハードウェア偏重が助長されるのは好ましくありません。 また、ソフトウェアの情報は、同一の研究分野内では相互リンクによって情報収集が可能ですが、分野を隔てた間での有用なソフトウェアの活用法7)8)9)10)について知るのは易しくありません。膨大なリンク集は、利用者の意向に沿った分類がなされているわけではなく、目的の情報をなかなか探し出せません。キーワード検索サイトでは、キーワードの選択が難しくヒットした中にから必要な情報だけを見つけ出すのは難しいものです。 そのため、新たな情報収集検索システムが必要と考えられますが、人と人との直接の情報交換に勝るものはありません。また、情報は個人的にも日本という単位でも入超にならないように提供して返すことも重要です。ソフトウェアの提供者となることは難しいですが、本稿で述べた内容と同程度のことを行う方法や組み合わせは他にもたくさんあります。もっと手軽で便利で安価な方法がありましたら、是非WWWなどで広く公開下さい。
1)Elliot Soloway, "No One Is Making Money In Educational Software"; Communications of The ACM, Vol.41, No.2, pp.11-15(1998)
翻訳は、"教育用のソフトウェアは儲からない"; 情報処理, Vol.39, No.7, pp.646-649 (1998)
2)立花 宏, "インターネット上の化学工学関連情報"; 分離技術, No.1, pp.23-29(1996) http://www-b.indchem.metro-u.ac.jp/SSPEJ/IN4CEs.html
3)化学関連サイトへのリンク http://www.nifty.ne.jp/forum/fchem/link.htm
4)Yahoo! Science:Chemistry http://www.yahoo.com/Science/Chemistry/
5)3D Molecular Modeling HomePage 東京薬科大学薬学部第二薬化学教室http://www.ps.toyaku.ac.jp/~dobashi/
6)生活環境化学の部屋 home page県立新潟女子短大生活科学科本間研究室 http://www.nicol.ac.jp/~honma/
7)Chemistry Tutorial and Resource Page http://www.mc.maricopa.edu/academic/phy_sci/Chemistry/faculty/dorland/chime.html
8)Macintosh The Chemical Tune-Up http://www.asahi-net.or.jp/~vp7m-ski/index.html
9)計算生化学http://www.biwa.or.jp/~k-sugino/
10)Macで蛋白質を見ようhttp://art.osaka-med.ac.jp/~med012/HomePage/Display/Display.html
他の関連文献については、こちらを御参照下さい。
 トップへ
トップへ  v2n1目次へ
v2n1目次へ
化学教育ジャーナルホームページへ

{kind=link}
{kind=link}
{kind=link}
{kind=link}